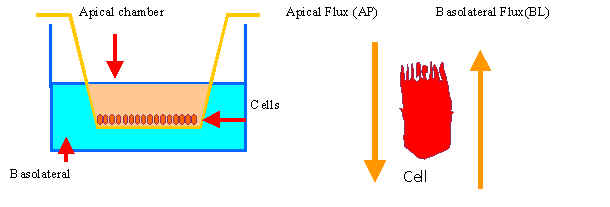
Клетки Caco-2 при культивировании в обычных для культур условиях образовывают монослой. Показано, что степень проницаемости (permeability rate) хорошо коррелирует со всасыванием веществ различной природы (в частности это показано на бетоблокаторах).
| Физико-химические свойства | Методы разделения | Стабильность |
| pKa (константа ионизации) | Количественное определение: | Гидролиз |
| lg P (октанол/вода) | ВЭЖХ | Окисление |
| lg D (липофильность) | ГЖХ | Светочувствительность |
| Расворимость (субстанция) | СКЖХ | Стабильность при хранении: |
| KIMM (константа иммобилизации на модельной системе) | валидизация | ускоренное |
| Тесты на распадаемость и растворимость (лек. форма) |
Идентификация метаболитов и примесей: |
долгосрочное |
| BCS | масс спектрометрия | |
| ИК | ||
| атомно-эмиссионный детектор |
В последнее время достаточно широкое распространение получило моделирование пассивного транспорта через эпителий кишечника. В качестве модели используется культура клеток Caco-2. Эти клетки получают из колонректальной карциносмы.
Клетки Caco-2 при культивировании в обычных для культур условиях образовывают монослой. Показано, что степень проницаемости (permeability rate) хорошо коррелирует со всасыванием веществ различной природы (в частности это показано на бетоблокаторах).
Для контроля качества фармацевтических продуктов используются тесты на распадаемость таблеток и на высвобождение (растворимость) действующего начала. Тест на растворимость следует признать более предпочтительным, поскольку абсорбция препарата в кровь из быстро распадающейся таблетки может быть медленной в тех случаях, если будет низкой скорость высвобождения препарата из таблетки, а после ее распада ѕ из гранул или их агрегатов.
Время распадаемости определяют по времени прохождения содержимого таблетки через специальное вибрирующее сито в жидкости определенного состава.
Для изучения растворимости в Фармакопее США предложены два утвержденных устройства ѕ «вращающаяся корзинка» и «лопасть для перемешивания». Первый из этих тестов утвержден для применения в России. Среди многочисленных факторов, влияющих на растворимость лекарственной формы следует отметить следующие: среда растворения (pH, ионная сила, вязкость), скорость вращения ротора, геометрическая форма устройства и температура. В качестве среды используется вода, искусственный желудочный сок и 0,1 н. HCl. Температура обычно равняется 37°С. Тесты растворимости, проводимые in vitro, оказываются чрезвычайно полезными при определении контроля качества при сравнении разных серий одной и той же лекарственной формы.
Простота и дешевизна тестов in vitro, позволяет их рассматривать как альтернативу сложным, дорогостоящим и этически небезупречным исследованиям по изучению биодоступности препарата in vivo. В связи с вышесказанным возникает проблема выявления корреляции между результатами экспериментов, проводимых in vitro и in vivo.
Как правило, рассматриваются корреляционные соотношения между параметрами теста растворимости (процент вещества, растворившегося за определенный промежуток времени, или, наоборот, время, необходимое для растворения определенного процента вещества) и параметрами фармакокинетики. Среди используемых in vivo параметров следует упомянуть Cmax, AUC, MRT, среднюю концентрацию вещества в плазме спустя 0,5 или 1 час после введения, максимальную скорость экскреции и кумулятивную экскрецию препарата за определенный промежуток времени. Согласно такому авторитету как Wagner (1970), наиболее предпочтительно использовать в качестве показателя in vitro величину времени, за которое растворяется 50% действующего вещества, а in vivo ѕ время полуабсорбции препарата.
Изучению корреляционных соотношений in vitro / in vivo посвящено достаточно большое количество публикаций. В некоторых случаях удается выявить достоверные корреляции in vitro / in vivo; во многих других случаях подобные соотношения не были выявлены или слабо коррелировали между собой. С одной стороны, несмотря на явные различия в скорости высвобождения in vitro, значимых различий между параметрами биодоступности сравниваемых лекарственных форм не было (Ylitalo, Lundell, 1975; Stoiropoulus et al., 1981; Hartley at al., 1991); в то же время одинаковые показатели теста на растворимость не обусловливали биоэквивалентность воспроизводимых препаратов (Banakar,1992).
| Группа | Растворимость | Проницаемость | Корреляция in vivo/in vitro |
| I | Высокая | Высокая |
Наблюдается в том случае, если скорость опорожнения желудка превышает скорость растворения |
| II | Низкая | Высокая | Выявлена при равенстве скорости растворимости in vivo и in vitro |
| III | Высокая | Низкая | Малая проницаемость мембран обусловливает скорость абсорбции |
| IV | Низкая | Низкая | Нет |
Биофармацевтическая классификация лекарственных веществ основывается на тестах растворимости. В настоящее время эта классификация принята FDA, как одно из оснований заменить изучение биодоступности какого-либо вещества на in vitro тест.
Согласно BCS препараты подразделены на 4 группы по степени их растворимости и проницаемости биомембран. Согласно этой классификации наибольший интерес в плане корреляций in vitro/in vivo представляют препараты II группы в которых скорость растворения вещества является лимитирующей стадией.
FDA определен список препаратов с различной степенью проницаемости через мембраны в качестве внутренних стандартов и маркеров для сравнительного исследования потенциальных лекарственных средств. Среди этих веществ в контексте дальнейших рассуждений отмечу, пропранолол или анаприлин, препарат с высокой степенью проницаемости.
| Препарат | Проницаемость | Примечание |
| альфа-метил-ДОФА | Низкая | Переносчик аминокислот |
| антипирин | Высокая | Маркер проницаемости |
| атенолол | Низкая | Внутренний стандарт |
| кофеин | Высокая | |
| карбамазепин | Высокая | |
| гидрохлортиазид | Низкая | IV группа |
| фуросемид | Низкая | IV группа |
| кетопрофен | Высокая | |
| маннитол | Вариабельная | Граничный маркер |
| метопролол | Высокая | Внутренний стандарт |
| напроксен | Высокая | |
| пропранолол | Высокая | |
| ранитидин | Низкая | Внутренний стандарт |
| теофилин | Высокая | |
| верапамил | Высокая | Маркер для P-гликопротеинов |
Согласно BCS тесты растворимости можно предложить в качестве изучения форм с контролируемым высвобождением.
Совместно с А.В. Авраменко нами было предпринято изучение фармакокинетики анаприлина, вводимого энтерально в формах с разной скоростью высвобождения и установление корреляции параметров фармакокинетики и скорости высвобождения in-vitro.
В качестве форм контролируемого высвобождения использовали пористые матричные системы (ПМС) с равномерным содержанием анаприлина. В качестве формы с быстрым высвобождением были взяты таблетки анаприлина производства ОАО “ХФК ”Акрихин” (Россия). Доза анаприлина во всех формах составляла 10 мг.
Пористые инертные матрицы готовили на основе сэвилена с добавкой аэросила для увеличения гидрофильности. В основе технологии изготовления матриц лежит фазоинверсионный термический процесс.. Изготовлены плоские матрицы толщиной 2,0 и 2,7 мм.
Проведено сравнительное изучение in vitro/in vivo ПМС двух размеров: №1- 3,0х2,7х5,0 мм (рабочая поверхность 2,7х5,0 мм); №2- 4,0х2,0х5,0 мм (рабочая поверхность 2,0х5,0 мм).
Кинетику высвобождения анаприлина из таблеток и ПМС двух размеров изучали на приборе “Вращающаяся корзинка”. Каждая форма изучалась в пяти опытах. В качестве среды высвобождения использовался 0,1 М раствор соляной кислоты, кислотность которого соответствует кислотности желудочного сока. Объем среды высвобождения в приборе “Вращающаяся корзинка” для каждого образца составлял 200 мл, скорость вращения корзинки- 60 оборотов в минуту. Объем проб, отбиравшихся для определения концентрации- 1 мл. Пробы отбирались через 2, 5, 10, 20, 40, 60 минут, далее с интервалом 30 минут до полного высвобождения вещества. После отбора пробы объем среды восстанавливался добавлением 1 мл чистой среды. Концентрацию анаприлина в среде определяли спектрофотометрически по собственному поглощению при длине волны 289 нм.
Экспериментальную фармакокинетику анаприлина исследовали на 15-ти кроликах-самцах породы “Шиншилла” массой 3,2-3,7 кг. Животные содержались в стандартных условиях вивария, при 12-часовом режиме освещения. Перед экспериментом за 18 часов и во время его животные голодали, получая только воду. Каждая форма была исследована на пяти животных.
Кровь из ушной вены отбирали непосредственно перед введением препарата и после него через 0,25, 0,5, 1,0, 2,0, 4,0, 8,0, 24,0 часа. Пробы крови выдерживали при комнатной температуре до образования сгустка, после чего образцы центрифугировали в течение 10 минут при n=2500 об./мин. в термостатируемой центрифуге при 00С и отбирали сыворотку. До анализа пробы сыворотки хранили при температуре -180 oС.
Анализ содержания анаприлина в сыворотке крови проводили с помощью высокоэффективной жидкостной хроматографии с флуоресцентной детекцией. Образцы сыворотки крови объемом 0,5 мл подщелачивали добавлением 200 мкл 10% раствора карбоната натрия. Экстракцию анаприлина проводили дважды, добавляя по 1,5 мл диэтилового эфира, встряхивая смесь в герметично закрытых пробирках в течение 5 мин. на продольном встряхивателе. Для лучшего разделения слоев образцы центрифугировали в течение 10 мин. при n=5000 об./мин. Эфирный экстракт отбирали, объединяли, упаривали досуха в токе азота на водяной бане при t=400С. Сухой остаток растворяли в 250 мкл метанола
Пробы хроматографировали на жидкостном хроматографе “Shimadzu LC-6A ” (Япония) в следующих условиях: хроматографическая колонка . Spherisorb С18 (250х4.6 мм), защищенная предколонкой, подвижная фаза 0,01М раствор калия дигидрофосфата и ацетонитрил в объемных соотношениях 45:55 (pH=3,0), скорость элюирования 2,0 мл/мин. Длина волны возбуждения- 280 нм, испускания- 340 нм. Объем вводимой пробы- 50 мкл. Время удерживания вещества составило 5,2±0,1 мин. Обработка хроматографических данных проводилась на компьютерном интеграторе по методу абсолютной калибровки. Предел обнаружения равнялся 3 нг/мл.
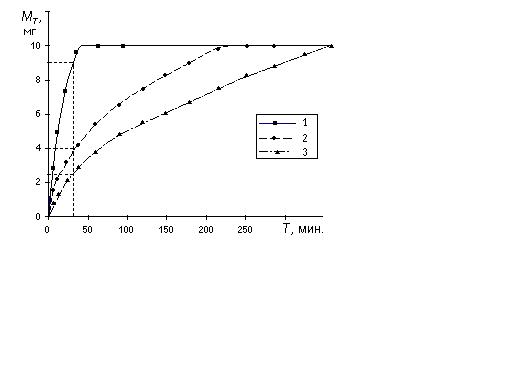
Усредненные данные о кинетике высвобождения анаприлина in-vitro из трех форм представлена на рисунке 2.
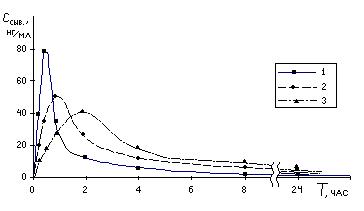
Как видно на рис. 3., при использовании систем контролируемого высвобождения достигается замедление всасывания, сглаживание пика концентрации и замедление выведения анаприлина.
Значения фармакокинетических параметров анаприлина приведены в нижеследующей таблице.
| № | Форма | M30, % | сmax, нг/мл | tmax, ч | MRT |
| 1 | Таблетка | 90±6 | 79,2±22,4 | 0,4±0,1 | 0,9±0,2 |
| 2 | ПМС №1 | 40±3 | 50,6*±15,7 | 1,1±0,3 | 2,0±0,3 |
| 3 | ПМС №2 | 25±2 | 40,2*±12,1 | 2,0±0,5 | 3,2±0,4 |
Анаприлин характеризуется высоким уровнем пресистемного метаболизма и широкой вариабельностью фармакокинетики, особенно по параметру сmax. Несмотря на это, по всем рассмотренным параметрам в разных группах, кроме сmax во 2-й и 3-й группах, установлены статистически достоверные отличия.
Установлено, что максимальная концентрация анаприлина в сыворотке крови прямо пропорциональна, а время максимума концентрации и среднее время удерживания вещества в организме обратно пропорциональны скорости высвобождения in-vitro. Уровень линейной корреляции параметров (величина R2) повышается в ряду сmax, tmax, MRT. В случае MRT R2 =0.83. е о корреляции позволяют прогнозировать экспериментальный фармакокинетический профиль анаприлина, вводимого энтерально в пористых матричных системах контролируемого высвобождения, исходя из скорости его высвобождения in-vitro.
Необходимо подчеркнуть, что установленные зависимости действуют только для систем контролируемого высвобождения с неравномерной скоростью высвобождения in-vitro, кинетический профиль которых подобен профилям исследованных систем. При переходе к системам, имеющим отличный от исследованных профиль высвобождения, например с постоянной скоростью высвобождения, зависимости между параметрами in-vitro и in-vivo могут измениться.
Таким образом, в результате проделанной работы установлены математические зависимости между параметрами экспериментальной фармакокинетики анаприлина и скоростью его высвобождения in-vitro. Полученные данные свидетельствуют о целесообразности использования систем контролируемого высвобождения анаприлина для достижения требуемой фармакокинетики.